
Chapitre 2 Lésions élémentaires des cellules, tissus et organes
Auteurs : Jean-baptiste Gibier et Morgane Stichelbout
Plan du chapitre
• Pré-requis
• Adaptation cellulaire et tissulaire
• Accumulation de substances intracellulaires
• Accumulation de substances intercellulaires
• Dégénérescence et mort cellulaire
• Savoir définir les termes suivants : homéostasie, lésion, adaptation cellulaire.
• Savoir définir et donner des exemples pour les termes suivants : atrophie, hypertrophie, aplasie, hypoplasie, hyperplasie, métaplasie.
• Savoir définir et donner des exemples de nécrose.
• Savoir définir et donner des exemples d'apoptose.
• Savoir définir la stéatose. Connaître les aspects macroscopiques et microscopiques de la stéatose.
• Savoir définir la cholestase. Connaître les aspects macroscopiques, microscopiques et les causes de la cholestase.
• Savoir donner des exemples de calcifications.
• Savoir définir l'hémosidérose. Connaître les caractéristiques microscopiques et les étiologies principales de l'hémosidérose.
• Savoir définir l'amylose. Connaître les caractéristiques microscopiques et les différents types d'amylose.
Pré-requis
Le travail d'un pathologiste débute par la description précise des lésions élémentaires. Ces lésions sont définies comme l'ensemble des altérations morphologiques présentes dans une structure et accessibles à l'observation. Elles peuvent être observables à différentes échelles et par différents moyens :
• macroscopiquement, lorsqu'elles sont visibles à l'échelle de l'organe ;
• en microscopie photonique, lorsqu'elles sont visibles à l'échelle du tissu et de la cellule ;
• en microscopie électronique, lorsqu'elles sont visibles à l'échelle des organites.
Le second temps du travail du pathologiste est de contextualiser ces lésions élémentaires. Ce travail est indispensable car la présence d'une lésion morphologique ne traduit pas forcément la présence d'une anomalie clinique. On attend du pathologiste qu'il distingue les lésions constituées témoignant d'une réaction adaptée de l'organisme ; des lésions évolutives témoignant d'une incapacité des cellules à fonctionner normalement.
Pour répondre à cette question, le pathologiste doit connaître les processus d'adaptation cellulaire et tissulaire, pouvant survenir aux différentes étapes du développement et de la vie, et qui aboutissent à la mise en place d'un nouvel état stable, à un changement de l'homéostasie tissulaire.
Le pathologiste doit aussi savoir reconnaitre quand ces mécanismes d'adaptation dépassent les capacités cellulaires ce qui se traduit le plus souvent par une mort cellulaire et une altération de la fonction de l'organe.
Dans certains cas, une altération morphologique peut orienter directement sur la nature de l'agression mais le plus souvent les mêmes lésions élémentaires peuvent s'observer dans des contextes extrêmement variés, ce qui souligne l'importance pour le pathologiste d'avoir accès à des informations cliniques précises pour faire ce travail de synthèse.
Adaptation cellulaire et tissulaire
Les cellules ont des capacités adaptatives pour faire face à une agression ou à un changement dans leur environnement. Certaines de ces modifications sont associées à des changements dans le nombre ou dans la taille des cellules et/ou de leurs constituants. Elles peuvent être irréversibles ou réversibles à l'arrêt de la cause.
Les principales réponses adaptatives d'une cellule et d'un tissu sont l'atrophie (ou hypotrophie), l'hypertrophie, l'aplasie, l'hypoplasie et l'hyperplasie et la métaplasie.
Attention : une même modification structurelle peut être observée dans un contexte physiologique ou en réponse à une condition pathologique (exemple : l'hypoplasie médullaire peut traduire un âge avancé ou bien être secondaire à une pathologie chez le sujet jeune).
Atrophie (hypotrophie)
Définitions
L'atrophie cellulaire est la diminution de la masse fonctionnelle d'une cellule habituellement liée à une diminution de son activité.
Elle se traduit par une diminution du volume cellulaire en rapport avec une diminution du nombre et de la taille des constituants normaux de la cellule.
À l'échelle tissulaire, l'atrophie peut être liée à l'atrophie cellulaire et/ou à la diminution du nombre des cellules.
Exemples
• Contexte physiologique : elle est liée le plus souvent à une involution hormonale : atrophie du thymus après la puberté, atrophie des ovaires et de l'endomètre après la ménopause.
• Contexte pathologique : une atrophie musculaire peut être observée après dénervation, ou lors de l'immobilisation prolongée d'un membre. Une atrophie tubulaire est fréquemment observée dans les zones de fibrose rénale (figure 02.01).
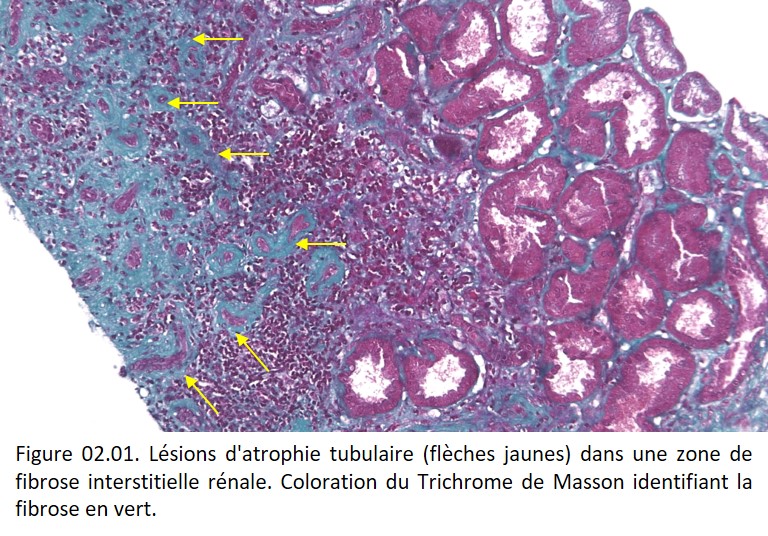{kind=link}
Hypertrophie
Définitions
L'hypertrophie cellulaire est une augmentation réversible de la taille d'une cellule en rapport avec une augmentation de la taille et du nombre de ses constituants. Cette hypertrophie va habituellement de pair avec une augmentation des stimuli et de l'activité de la cellule.
L'hypertrophie relève de deux mécanismes principaux :
• Une augmentation de l'activité mécanique ou métabolique de la cellule ;
• une stimulation hormonale accrue.
À l'échelle tissulaire, l'hypertrophie est une augmentation du volume d'un tissu ou d'un organe liée à une hypertrophie cellulaire et/ou à une augmentation du nombre de cellules.
Attention : une augmentation de la taille ou du poids d'un organe peuvent être secondaires à d'autres mécanismes comme par exemple à la dilatation des cavités d'un organe ou bien à l'infiltration du tissu par de la fibrose, de l'œdème ou une substance anormale. Ces modifications du tissu interstitiel peuvent masquer une réelle atrophie (ex. : lipomatose musculaire).
Exemples
• Contexte physiologique : hypertrophie des muscles squelettiques du sportif ; hypertrophie musculaire lisse du myomètre au cours de la grossesse sous l'effet des estrogènes.
• Contexte pathologique : hypertrophie thyroïdienne par hypersécrétion d'hormone thyréotrope.
Aplasie et hypoplasie
Définitions
L'aplasie est l'absence d'un organe provoquée par l'absence de développement de son ébauche embryonnaire, et par extension, l'arrêt transitoire ou définitif de la multiplication cellulaire dans un tissu qui devrait normalement se renouveler en permanence.
L'hypoplasie est un développement embryologique anormal d'un viscère ou d'une partie d'un viscère aboutissant à un organe fonctionnel mais trop petit, et par extension le développement insuffisant d'un tissu lorsque les stimuli assurant sa trophicité normale diminuent ou cessent.
Attention : l'aplasie ne doit pas être confondue avec l'agénésie. L'agénésie désigne l'absence complète d'un organe en raison de l'absence d'ébauche embryonnaire.
Exemples
• Contexte physiologique : hypoplasie endométriale et testiculaire au cours de la sénescence.
• Contexte pathologique : aplasie ou hypoplasie de la moelle hématopoïétique secondaire aux radiations ionisantes. Hypoplasie ou aplasie associées à des anomalies du développement (figure 02.02).
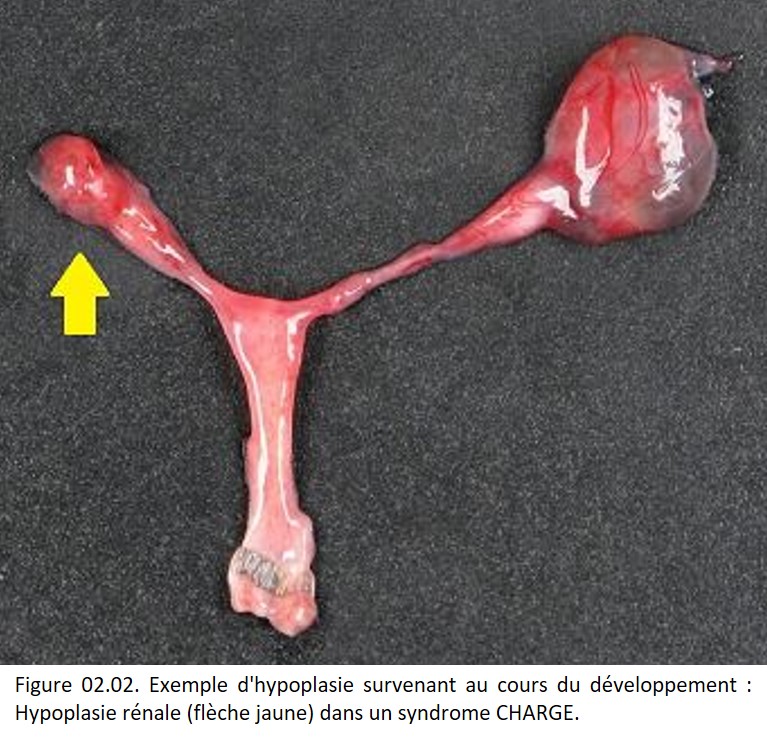{kind=link}
Hyperplasie
Définitions
L'hyperplasie est l'augmentation anormale du nombre de cellules d'un tissu ou d'un organe, sans modification de l'architecture, résultant habituellement en l'augmentation de volume du tissu ou de l'organe concerné. Elle est habituellement témoin d'une hyperactivité fonctionnelle.
Elle survient surtout dans les tissus capables de renouvellement (épiderme, épithélium intestinal, parenchyme hépatique) et ne s'observe pas dans les tissus à renouvellement lents ou stables (myocarde, muscle squelettique, tissu neuronal).
Elle est souvent associée à une hypertrophie cellulaire, avec laquelle elle partage des causes communes.
Exemples
• Contexte physiologique : hyperplasie mammaire par stimulation hormonale au cours de la grossesse.
• Contexte pathologique : hyperplasie surrénalienne au cours d'un hypercorticisme hypophysaire.
Métaplasie
Définition
La métaplasie est une anomalie acquise résultant de la transformation d'un tissu normal en un autre tissu normal, de structure et de fonctions différentes, normal quant à son architecture, mais anormal quant à sa localisation.
Elle intéresse surtout les tissus épithéliaux, particulièrement les muqueuses, et s'observe aussi dans les tissus conjonctifs. La métaplasie est qualifiée par un adjectif qui correspond à la nature du tissu résultant de la transformation.
Exemples
• Contexte physiologique : métaplasie déciduale du chorion cytogène de l'endomètre ;
• Contexte pathologique : une métaplasie intestinale de l'épithélium gastrique (Gastrite atrophique de Biermer) (figure 02.03) ; une métaplasie glandulaire d'un épithélium malpighien (œsophage de Barrett) (figure 02.04) ; une métaplasie malpighienne de l'épithélium glandulaire bronchique ; une métaplasie malpighienne de la muqueuse glandulaire endocervicale du col de l'utérus.
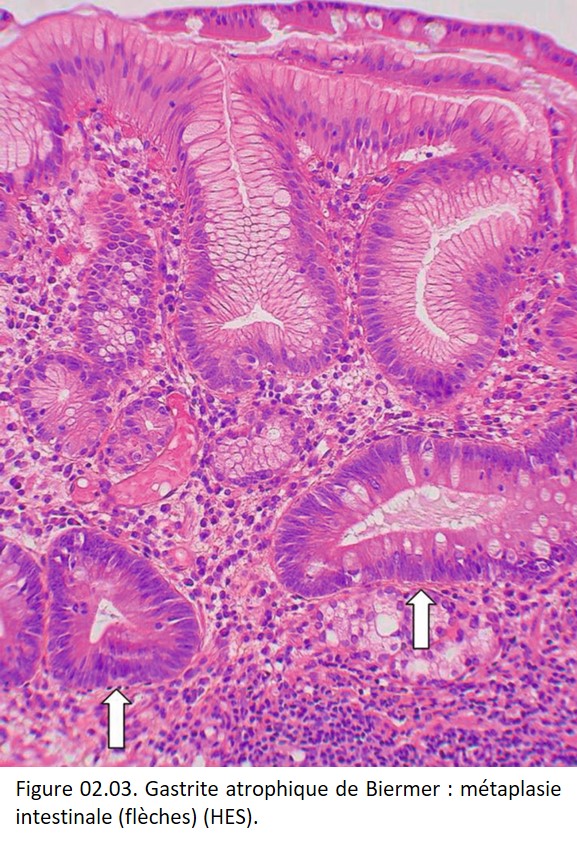{kind=link}
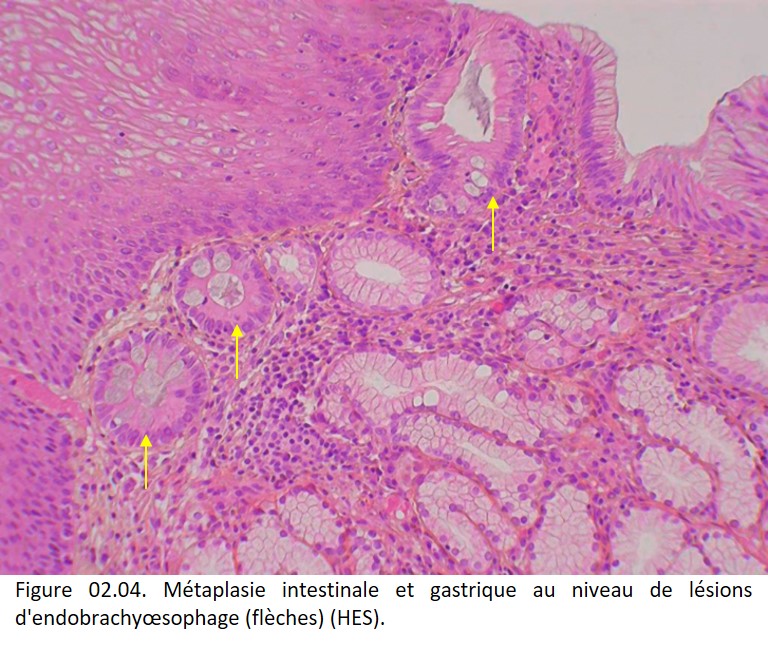{kind=link}
Terminologie utilisée en pathologie du développement
Au cours de la période prénatale, des agressions de nature variée peuvent aboutir à un arrêt total ou partiel du développement d'un organe ou même d'une partie du corps. Ces modifications sont congénitales, présentes dès la naissance. On distingue les malformations primaires résultant d'un événement génétiquement déterminé (intrinsèque) et les malformations secondaires résultant de la perturbation du développement normal par un facteur extrinsèque (déformation ou disruption). Pour décrire ces modifications propres à la pathologie fœtale, un lexique spécifique est mis à disposition du pathologiste. On distingue notamment quatre anomalies élémentaires définies de la manière suivante :
• malformation primaire : anomalie morphologique irréversible d'un organe, d'une partie d'un organe ou d'une plus grande partie du corps, qui résulte d'un processus intrinsèquement anormal du développement. Exemples : fente palatine, communication inter-ventriculaire ;
• disruption : anomalie morphologique d'un organe, d'une partie d'un organe ou d'une plus grande partie du corps, qui résulte d'une altération extrinsèque ou d'une interférence avec un processus initialement normal du développement. Exemples : amputations digitales dans une maladie des brides amniotiques, anomalies cérébrales en cas de toxoplasmose congénitale, malformations liées à des agents physiques comme les rayons ionisants, amputation transverse d'un membre d'origine vasculaire ;
• déformation : anomalie de forme, de taille ou de position d'une partie du corps, provoquée par des phénomènes mécaniques. Exemples : déformations des membres liées à un oligo-anamnios (ankylose et déformation des membres avec des pieds bots) ;
• dysplasie : organisation anormale des cellules dans un tissu et ses conséquences morphologiques (dyshistogénèse) à l'échelle macroscopique, microscopique et moléculaire. Exemple : dysplasie rénale multikystique (figure 02.05).
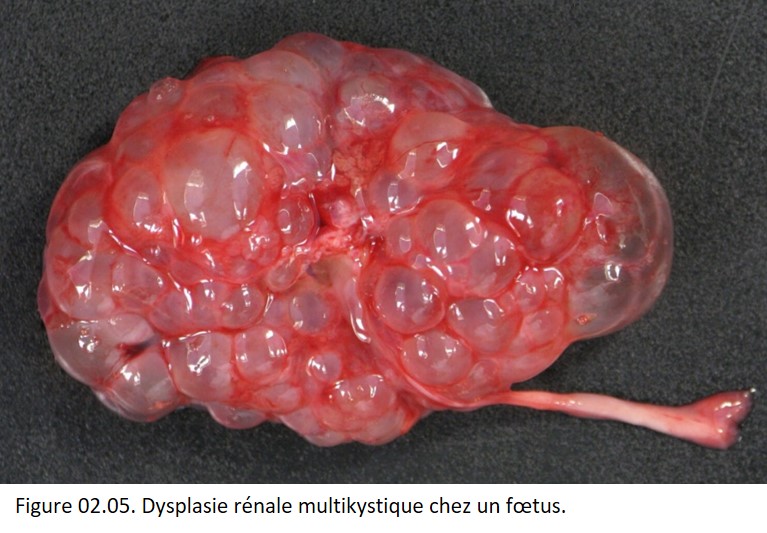{kind=link}
Accumulation de substances intracellulaires
Dans certaines situations, il existe une accumulation anormale de substances variées (lipides, protéines, pigments, cristaux) dans le cytoplasme des cellules.
Ces substances peuvent être produites par la cellule et on parle alors de substances d'origine endogène ; ou bien être étrangères à l'organisme et on parle alors de substances exogènes.
Les substances endogènes peuvent s'accumuler dans plusieurs circonstances :
• en cas de production excessive ;
• en cas de perturbation du métabolisme normal (exemple de la stéatose) ;
• en cas de défaut de transport hors de la cellule (exemple de la cholestase) ;
• lorsque la substance produite résiste aux mécanismes de dégradation (exemple des maladies lysosomales).
Souvent, c'est une combinaison de ces différentes situations qui est à l'origine de l'accumulation. Par exemple, lorsque les plasmocytes produisent beaucoup d'immunoglobulines et lorsqu'il existe une dérégulation des mécanismes de sécrétion, il est possible d'observer des agglomérats de ces protéines dans le cytoplasme ou dans le noyau des plasmocytes, agglomérats appelés corps de Dutcher (figure 02.06).
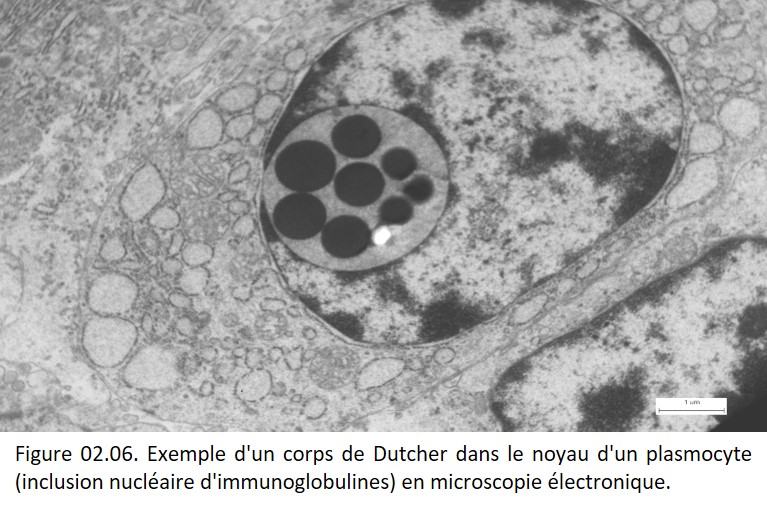{kind=link}
Les substances exogènes s'accumulent parfois car elles ne sont pas métabolisables et qu'elles résistent aux mécanismes de dégradation enzymatique. Elles vont donc persister au cours de la vie. Dans certains cas, ces substances vont entrainer une inflammation locale et favorisent l'émergence de certaines pathologies (c'est le cas de très nombreuses maladies professionnelles) alors que parfois elles sont relativement inertes (exemple : des pigments secondaires au tatouage).
Au même titre que les anomalies structurelles, l'accumulation de substance intracellulaire peut être irréversible ou transitoire et être secondaire à des conditions pathologiques ou physiologiques (exemple physiologique : accumulation de pigments de lipofuschine au cours du vieillissement).
Stéatose hépatocytaire
Définition
La stéatose ou dégénérescence graisseuse est l'accumulation anormale de triglycérides dans les cellules parenchymateuses. Elle est fréquemment observée dans les hépatocytes, fortement impliqués dans le métabolisme lipidique, on parle alors de stéatose hépatocytaire. Plus rarement la stéatose peut aussi s'observer dans d'autres organes (cœur, rein, muscle).
Causes
Elles sont multiples : toxiques (alcool, médicament), nutritionnelles, diabète, obésité, hypoxie, infection (hépatite virale C). Dans les pays développés, sa cause la plus fréquente est l'alcoolisme.
Physiopathologie
À l'état normal, les acides gras issus du tissu adipeux ou de l'alimentation sont transportés dans les hépatocytes, où ils sont estérifiés en triglycérides, puis convertis en cholestérol ou en phospholipides ou oxydés en corps cétoniques. D'autres acides gras peuvent être synthétisés à partir de l'acétate. La libération des triglycérides par les hépatocytes se fait sous forme de lipoprotéines après leur conjugaison à des apoprotéines.
L'accumulation de triglycérides peut être liée, selon l'étiologie, à une anomalie au niveau de chaque étape métabolique, depuis l'entrée des acides gras jusqu'à leur sortie sous forme de lipoprotéines : l'alcool est un toxique pour les fonctions mitochondriales et microsomales des hépatocytes, la malnutrition diminue les synthèses d'apoprotéines, l'anoxie inhibe l'oxydation des acides gras, et le jeûne en augmente la mobilisation périphérique.
Macroscopie
Dans les stéatoses importantes, le volume du foie est augmenté, sa consistance est molle, sa couleur est jaune, laissant à la coupe une marque de dépôts graisseux (figure 02.07).
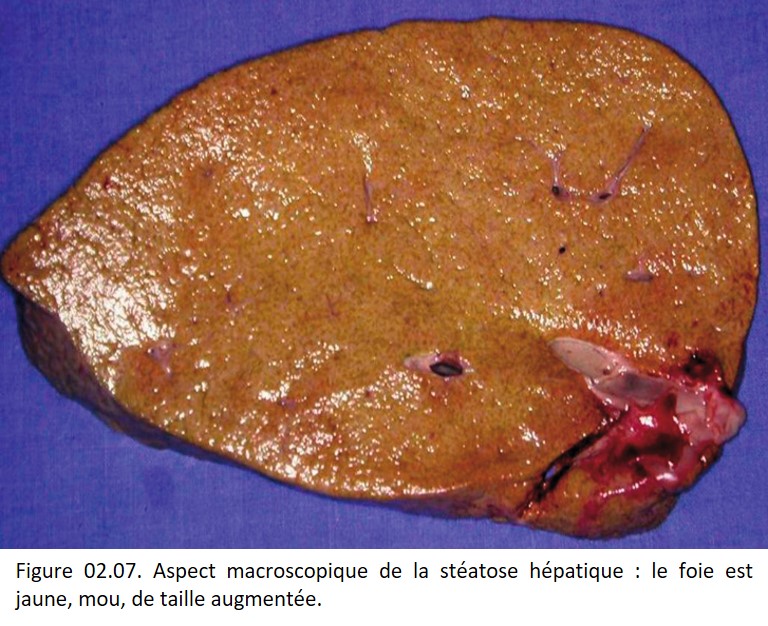{kind=link}
Histologie
Les cytoplasmes des hépatocytes contiennent des vacuoles optiquement vides si les prélèvements ont été colorés après inclusion en paraffine car les triglycérides contenus dans ces vacuoles ont été dissous lors du passage dans les solvants tels que le toluène ou le xylène. La stéatose peut ainsi se présenter sous deux formes :
• macrovacuolaire, la plus fréquente, où les gouttelettes de stéatose refoulent le noyau en périphérie de la cellule, et peuvent à l'extrême provoquer une rupture des membranes cellulaires et provoquer la formation de kystes graisseux (figure 02.08) ;
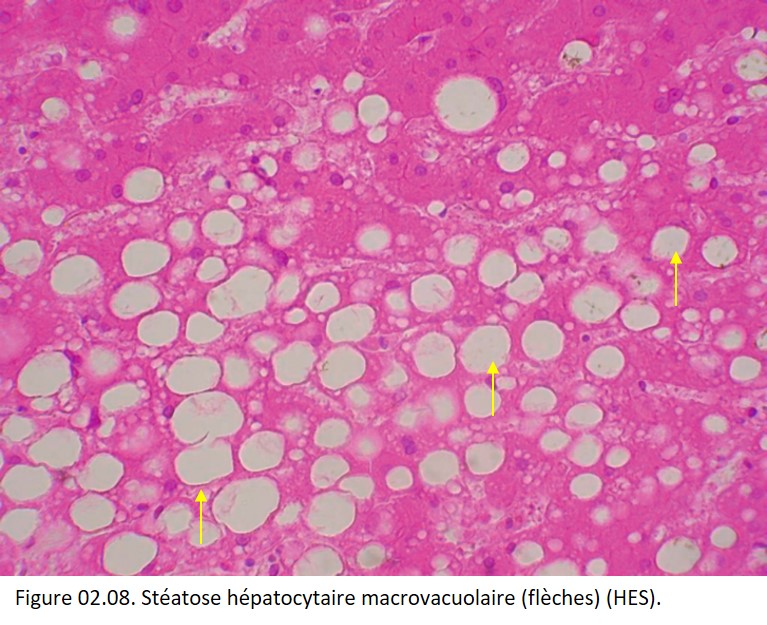{kind=link}
• microvacuolaire, rare, où le noyau reste central et où les vacuoles, très petites, peuvent être difficiles à voir. Certaines étiologies en sont plus volontiers à l'origine : stéatose aiguë gravidique, stéatose toxique médicamenteuse.
Pour visualiser les graisses directement, le prélèvement doit être congelé à l'état non fixé, puis coloré par des techniques spéciales comme le rouge à huile ou le noir Soudan.
La stéatose peut être répartie de façon aléatoire dans le parenchyme hépatique ou siéger préférentiellement dans un territoire fonctionnel, par exemple dans la région centrolobulaire en cas d'hypoxie ou d'intoxication.
Évolution
Les lésions de stéatose sont réversibles à l'arrêt de l'agression.
Cholestase
Définition
Une cholestase est définie histologiquement comme une accumulation visible de bile dans le tissu hépatique. La bile est un liquide aqueux majoritairement constitué d'acides biliaires, de cholestérol (tous deux produits par les hépatocytes) et de pigments biliaires dont la bilirubine (produits lors de la dégradation des globules rouges).
Attention : le terme cholestase signifie littéralement accumulation (ou « stase ») du « chole » c'est-à-dire de la bile. Toutefois, seuls les pigments biliaires sont observables directement sur une coloration standard de l'HES.
Physiopathologie et causes
Le maintien du flux biliaire est dépendant à la fois de la sécrétion des acides biliaires par les hépatocytes et de l'acheminement de la bile du foie jusqu'au duodénum par les voies biliaires intra et extra-hépatiques.
Une cholestase peut donc s'observer en cas :
• d'atteinte hépatocytaire (exemple : hépatite alcoolique, hépatite virale) ;
• d'atteinte des voies biliaires intra-hépatiques (exemple : cholangite lors d'une cirrhose biliaire primitive) ;
• d'obstruction des voies biliaires extra-hépatiques (exemples : adénocarcinome du pancréas, lithiase).
Macroscopie
La cholestase est responsable d'une coloration verte du foie (figure 02.09).
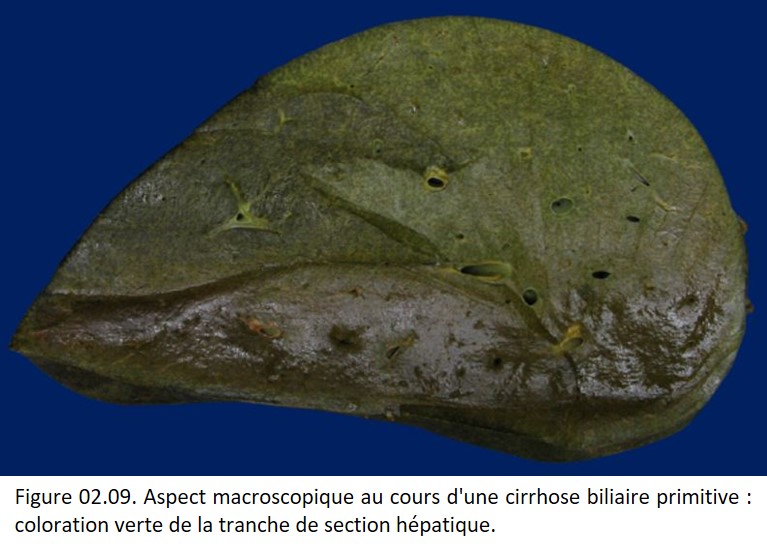{kind=link}
Histologie
La cholestase peut se manifester par des amas de bile de couleur brun verdâtre dans les canalicules inter hépatocytaires (figure 02.10) ou dans les canaux biliaires interlobulaires des espaces portes. La bile peut également siéger dans les hépatocytes et les macrophages et peut être difficile à différencier des lipofuschines.
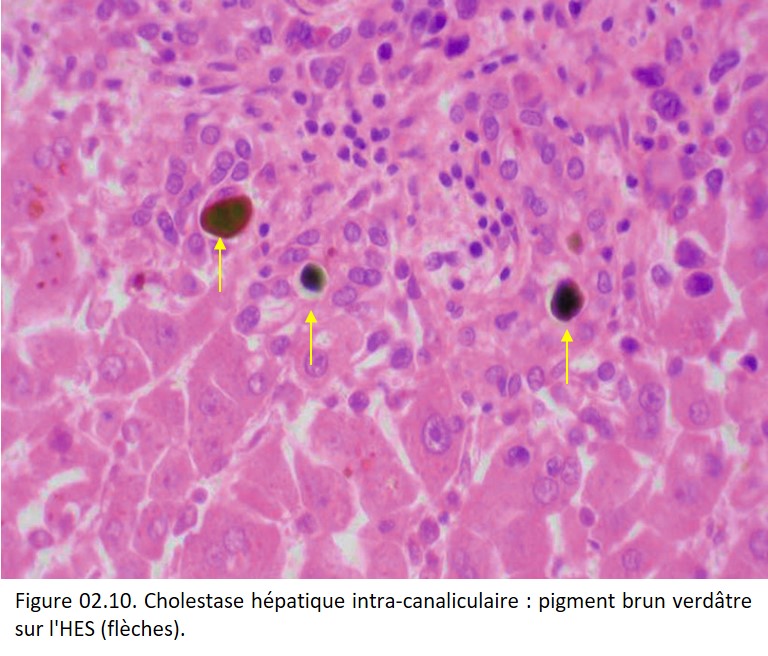{kind=link}
D'autres signes secondaires à l'accumulation des autres constituants de la bile peuvent être observés :
• la stase des acides biliaires entraine une souffrance hépatocytaire caractérisée par un gonflement et une clarification du cytoplasme ;
• la présence de cuivre, normalement secrété avec la bile, peut être mise en évidence dans les hépatocytes péri-portaux en utilisant la coloration de la rhodamine ;
• l'accumulation de cholestérol peut donner un aspect spumeux aux cellules.
Hémosidérose
L'hémosidérine est un pigment endogène brun jaunâtre qui dérive de l'hémoglobine. C'est une forme de stockage du fer dans les cellules. L'hémosidérine peut s'accumuler dans l'organisme, localement ou de façon diffuse. La surcharge peut être localisée (évolution d'une hémorragie par exemple) ou diffuse (anomalie génétique du métabolisme du fer par exemple).
On peut aussi observer, rarement, des surcharges en fer d'origine exogène, divers tissus pouvant être infiltrés de particules de fer exogène, par exemple le poumon chez des soudeurs à l'arc et les ouvriers des mines de fer. La sidérose pulmonaire est sans conséquences physiopathologiques mais elle est fréquemment associée à une surcharge en silice (silicose), présente aussi dans l'air inhalé.
Caractéristiques histologiques
Sur une coloration par l'HES, les amas d'hémosidérine suffisamment volumineux sont visibles sous l'aspect de granulations brun ocre un peu brillantes (figure 02.11).
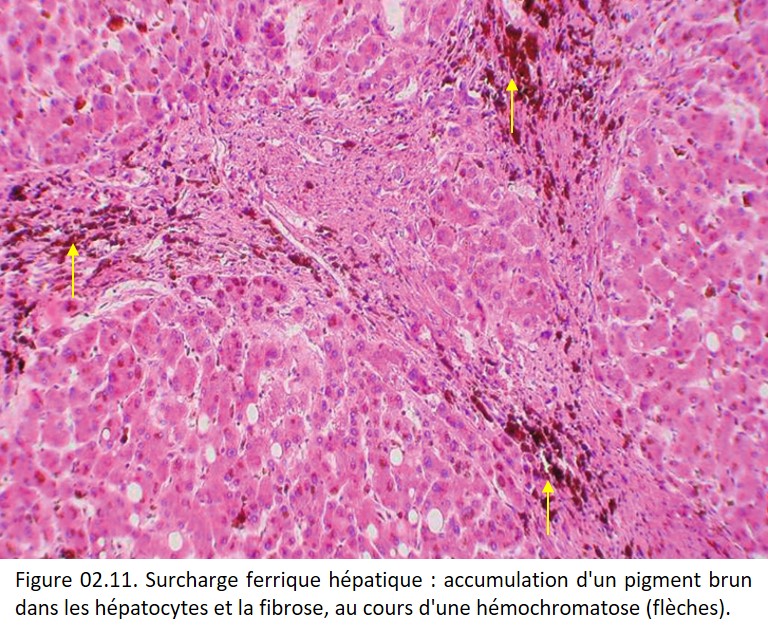{kind=link}
Des réactions sensibles et spécifiques permettent de caractériser le fer ionisé, la plus utilisée étant la réaction de Perls qui colore le fer ionisé en bleu (figure 02.12).
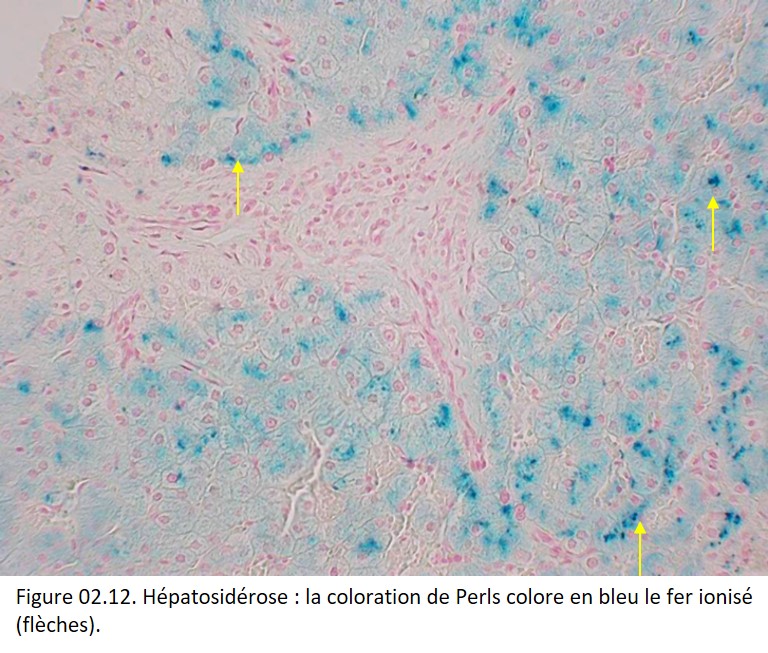{kind=link}
Hémosidérose localisée
L'accumulation locale de fer peut être liée à une hémorragie macroscopique ou de multiples hémorragies microscopiques.
Quand les hématies sont lysées, les lysosomes des macrophages transforment l'hémoglobine en hémosidérine, en passant par les diverses étapes de pigments intermédiaires (biliverdine, bilirubine), ce qui explique les variations de teinte lors de l'évolution d'un hématome.
Exemples : sidérose du « poumon cardiaque » ; cicatrices « tatouées » des infarctus hémorragiques (poumon) ; évolution des thromboses.
Hémosidérose généralisée
Elle correspond à une augmentation des réserves de fer de l'organisme, aboutissant à une surcharge polyviscérale. Le fer en excès s'accumule dans les macrophages et dans les cellules parenchymateuses. La surcharge peut être visible macroscopiquement si elle est importante, et se traduit alors par une coloration brune des viscères, voire une sensation de dureté et de crissement à la coupe (figure 02.13).
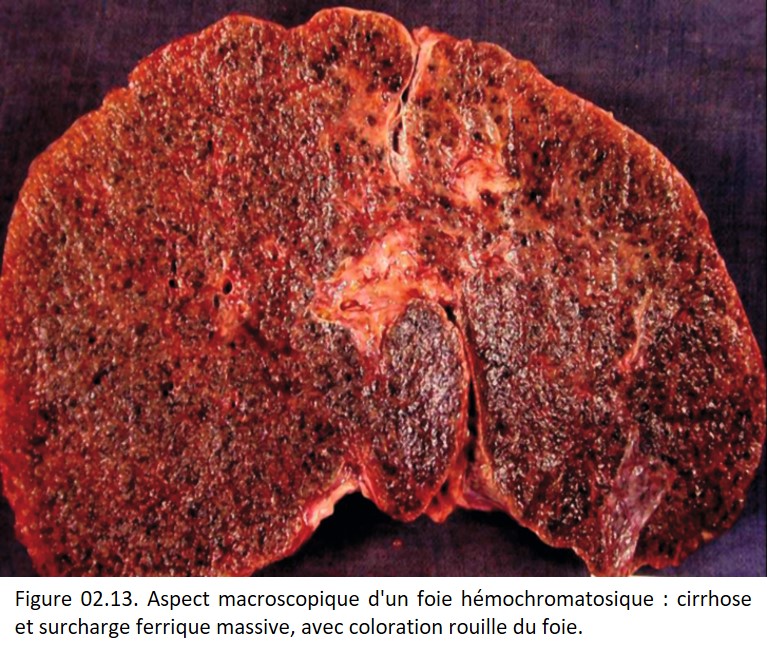{kind=link}
L'hémosidérose généralisée peut être primitive ou secondaire. Plusieurs mécanismes sont possibles :
• accroissement de l'absorption duodénale du fer alimentaire ;
• anomalie de l'utilisation du fer ;
• transfusions sanguines répétées.
Hémosidéroses généralisées secondaires
Il s'agit habituellement d'une hémosidérose pure, sans sclérose. Le fer s'accumule dans les phagocytes mononucléés du foie (cellules de Küpffer), de la rate, de la moelle osseuse, des ganglions lymphatiques et dans les macrophages dispersés dans d'autres organes (peau, pancréas, rein). Lorsque la surcharge augmente, les cellules parenchymateuses peuvent être atteintes (foie, pancréas, cœur, glandes endocrines). La localisation des dépôts peut varier en fonction du mécanisme en cause.
Hémosidérose généralisée primitive ou hémochromatose
C'est une maladie héréditaire à transmission autosomique récessive. L'accumulation de fer dans les cellules parenchymateuses aboutit à leur destruction et à une fibrose, en particulier le foie, le pancréas, le cœur, et les glandes endocrines. Les manifestations cliniques résultent surtout de l'atteinte de ces organes. Elles apparaissent pour un stock de fer de 30 à 50 g (10 fois le stock normal). Les patients ont un risque augmenté de développer un carcinome hépatocellulaire.
Silicose
Définition
La silicose est une pneumoconiose fibrosante provoquée par l'inhalation de particules de poussières de silice (silice cristalline). Les pneumoconioses sont des affections pulmonaires caractérisées par des dépôts de poussières inorganiques (minérales ou métalliques) dans le tissu pulmonaire.
Physiopathologie
Lorsque les poussières de silice atteignent les bronches, des macrophages alvéolaires vont phagocyter ces particules afin de les détruire. Toutefois, ils ne disposent pas du matériel enzymatique nécessaire à la dégradation de ces particules inorganiques.
La persistance de ces particules dans le cytoplasme des macrophages va conduire à la libération de signaux pro-inflammatoires, pro-fibrosants puis à la mort des macrophages. La silice est alors libérée dans le tissu interstitiel autour des petites bronchioles où elle entraine la formation des nodules silicotiques.
Histologie
La fibrose est le plus souvent localisée dans les zones où les dépôts de particules sont les plus importants et prédomine donc au sommet des poumons, dans les territoires péri-bronchiolaires centro-acinaires ou sous-pleuraux en formant des nodules caractéristiques.
Ces nodules d'abord principalement constitués de macrophages contenant des particules biréfringentes en lumière polarisée (talc, mica et silicates) sont progressivement remplacés par du collagène. Dans les lésions anciennes, ces nodules sont acellulaires et uniquement constitués d'une fibrose hyaline à disposition « tourbillonnante » caractéristique (figure 02.14). En général, il s'y associe une importante anthracose liée à l'accumulation de particules de carbone et de suie et responsable d'un aspect noir du poumon.
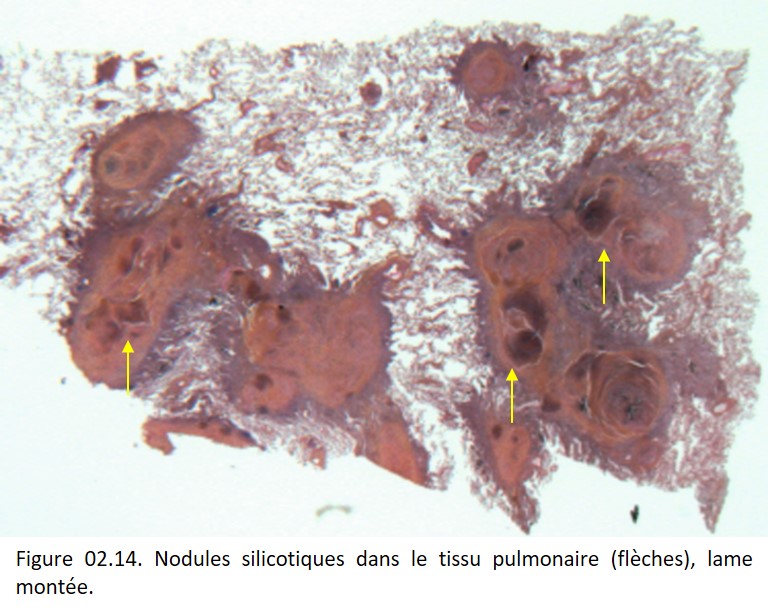{kind=link}
Évolution
En cas d'exposition massive (rare), il est possible d'observer des manifestations aiguës de la maladie qui peuvent être très sévères.
Le plus souvent, les patients atteints de silicose évoluent lentement vers l'insuffisance respiratoire chronique. Ils présentent un risque accru d'infection à mycobactéries et sont également à risque de développer un cancer bronchique. Cette complication est reconnue comme cette maladie professionnelle.
Accumulation de substances intercellulaires
À l'instar de ce qui se produit dans les cellules, le compartiment intercellulaire peut être le siège de modifications lors d'une agression.
Parfois, la réponse de l'organisme est caractérisée par l'accumulation de certains composés normalement présents dans le tissu interstitiel (exemple de la fibrose ou de l'œdème) et plus rarement par la formation d'une substance normalement absente des tissus (exemple de l'amylose).
Il faut noter que dans certains cas, l'accumulation d'une substance est d'abord d'origine intracellulaire avant que ces effets se manifestent dans le compartiment intercellulaire. C'est par exemple le cas dans la cholestase ou dans la silicose qui ont été abordées au chapitre précédent.
Amylose
L'amylose désigne une famille de maladies qui ont en commun la formation extracellulaire d'une substance protéique amorphe (la substance amyloïde) ayant des caractéristiques optiques spécifiques : elle est colorée en rouge sur la coloration du rouge Congo et elle présente une biréfringence jaune-vert lorsqu'elle est étudiée en lumière polarisée (figure 02.15).
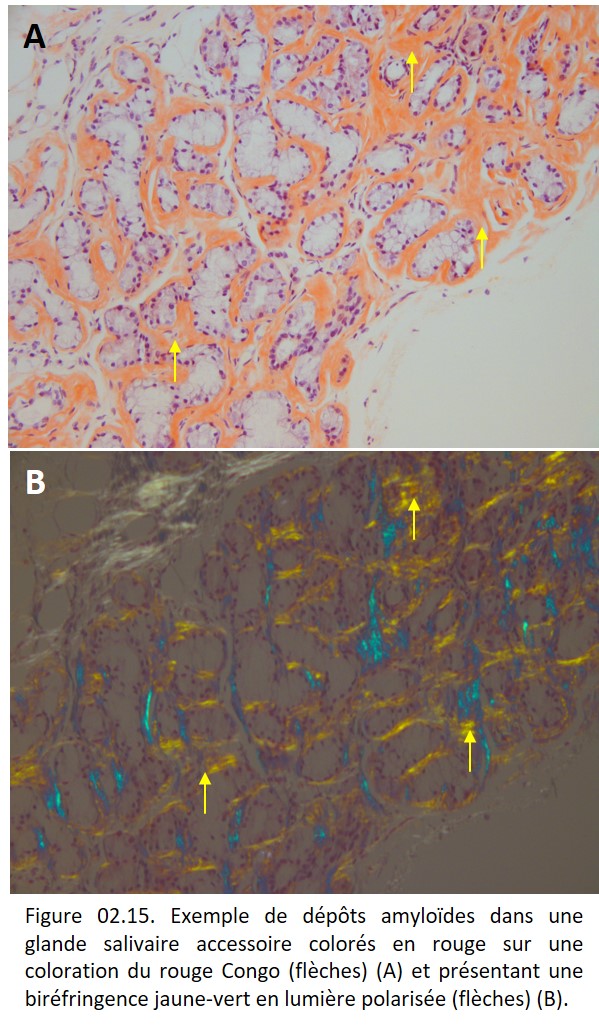{kind=link}
Les propriétés tinctoriales propres à la substance amyloïde sont secondaires à la nature structurelle des dépôts. En effet, quel que soit le type d'amylose, les protéines pathologiques s'agrègent sous forme hautement organisée en feuillets β plissés antiparallèles formant des fibrilles insolubles de 7 à 10 nm de diamètre, disposées en tous sens.
Physiopathologie
Les mécanismes de la formation des fibrilles amyloïdes sont complexes. La plupart des protéines amyloïdogènes ont une certaine susceptibilité à former des fibrilles dans des conditions biochimiques particulières, mais les déterminants exacts de leur formation in vivo sont méconnus. Deux mécanismes principaux peuvent être distingués selon que l'amylose est secondaire :
• à la production d'une protéine mutée. Dans ces cas, une mutation a rendu la protéine instable ce qui augmente son pouvoir amyloïdogène. C'est le cas des amyloses AL et des amyloses héréditaires ;
• à la production d'une protéine non mutée. C'est le cas des amyloses survenant au cours du vieillissement comme l'amylose cardiaque à transthyrétine ou de certaines amyloses cérébrales.
Composition et classification des amyloses
Au sein des dépôts amyloïdes, un certain nombre de constituants sont communs à tous les types d'amylose mais en proportion variable. Ils définissent ce que l'on appelle la « signature amyloïde ». Il s'agit notamment de certains composants de la matrice extracellulaire (comme les protéoglycanes), de lipoprotéines (Apolipoprotéine E) et du composant amyloïde P.
Seule l'identification de la protéine amyloïde, c'est-à-dire de la protéine qui est responsable de la formation de la substance amyloïde, permet de donner le type exact de l'amylose.
Dans la classification actuelle, 36 protéines potentiellement amyloïdes ont été identifiées chez l'homme.
Principaux types d'amylose en France
Si l'on connait plus de 30 types différents d'amylose, il s'agit souvent de pathologies héréditaires extrêmement rares et, en pratique, trois types d'amylose représentent plus de 90 % des cas d'amylose en France.
L'amylose AL
L'amylose AL est secondaire à l'agrégation dans les tissus de chaînes légères monoclonales d'immunoglobuline. Elle est donc secondaire à l'existence d'une prolifération clonale lympho-plasmocytaire. Cette dernière peut être à un stade avancé et cliniquement parlante comme lorsque l'amylose AL survient au cours d'un myélome ou d'une maladie de Waldenström ou bien au contraire l'amylose AL peut être la seule manifestation associée à ce clone tumoral.
L'amylose à transthyrétine (ATTR)
C'est une amylose de découverte plus récente mais dont l'incidence ne cesse d'augmenter en raison d'une meilleure connaissance et d'un meilleur dépistage de cette maladie. Elle regroupe deux types très différents de maladie :
• amylose à transthyrétine mutée : c'est la plus fréquente des amyloses héréditaires en France. Elle a été décrite initialement dans des familles de patients d'origine portugaise. Elle est essentiellement responsable d'une polyneuropathie sensitive et d'une atteinte cardiaque et plus rarement rénale ;
• amylose à transthyrétine non mutée ou amylose sénile : cette forme d'amylose touche essentiellement le cœur et survient chez les personnes âgées.
L'amylose AA
La protéine SAA produite en excès lors des situations inflammatoires chroniques est responsable de ce type d'amylose. Elle est de plus en plus rare dans les pays occidentaux en raison notamment d'une meilleure prise en charge des maladies infectieuses. Les principales maladies à l'origine de l'amylose AA sont les rhumatismes inflammatoires chroniques, les infections chroniques, les maladies inflammatoires chroniques de l'intestin (MICI) et les maladies auto-inflammatoires dont la fièvre méditerranéenne familiale. Il est à noter que l'obésité est une nouvelle cause environnementale d'amylose AA.
Attention : la fréquence respective des différents types d'amylose varie selon l'atteinte d'organe et de l'origine ethnique des patients. Par exemple, l'amylose ALECT2 est exceptionnelle chez les patients caucasiens mais est la deuxième cause d'amylose rénale chez les patients d'origine mexicaine aux États-Unis.
Atteinte d'organe et tropisme tissulaire
Le plus souvent, les amyloses sont des maladies systémiques qui peuvent toucher tous les organes. Certains types d'amylose intéressent plus souvent un organe en particulier (exemple de l'amylose à fibrinogène qui s'observe essentiellement dans le rein). Les déterminants du tropisme tissulaire des dépôts amyloïdes sont encore mal élucidés à ce jour.
Dans certains cas, les amyloses peuvent rester localisées à un organe ou à une région de l'organisme. On parle alors d'amylose localisée. Ces formes localisées d'amylose sont souvent liées à une amylose AL lorsqu'elle touche la région ORL, le poumon ou encore les os/tissus mous.
Macroscopie
La présence de dépôts amyloïdes s'accompagne, lorsque les dépôts sont abondants, d'une augmentation de volume de l'organe lésé (foie, rate, rein, cœur, langue). Ils peuvent être inapparents, ou responsables d'une infiltration cireuse et ferme, nodulaire ou diffuse, selon les organes et le siège des dépôts. Dans la rate par exemple, l'atteinte peut être nodulaire en « grains de tapioca » (rate sagou) ou diffuse, lardacée.
Histologie et typage
Les dépôts amyloïdes sont amorphes et de topographie majoritairement extracellulaire. Par définition, ils sont colorés par le rouge Congo et présentent une biréfringence jaune-vert en lumière polarisée.
Au-delà de la détection de l'amylose dans le tissu, l'objectif pour le pathologiste est de déterminer le type de l'amylose.
Pour mener à bien cette étape de typage, le pathologiste dispose de plusieurs outils :
• L'analyse du mode de dépot dans les tissus : dans certains cas, le mode de dépot des dépôts amyloïdes dans les tissus permet d'orienter le type d'amylose (figure 02.16).
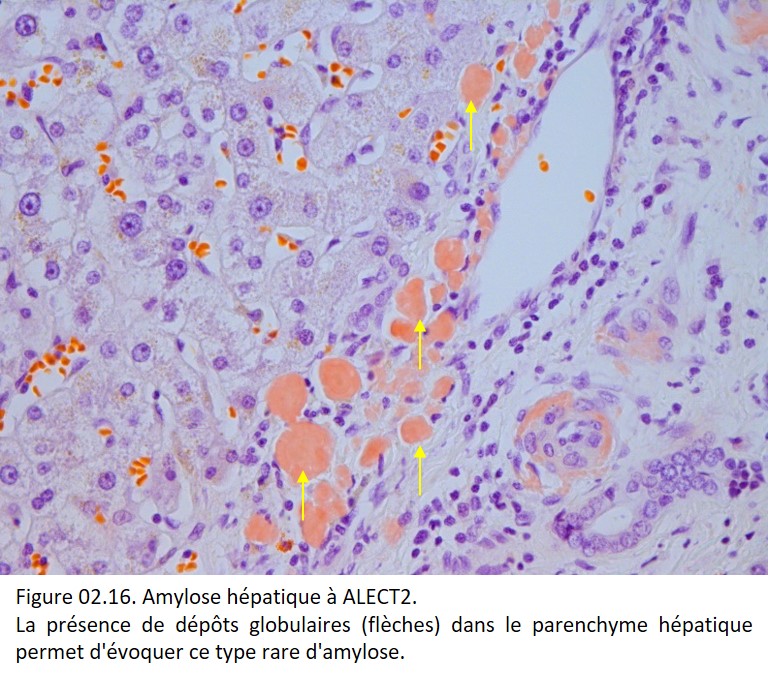{kind=link}
• L'immunohistochimie et l'immunofluorescence : ces techniques permettent la mise en évidence de la protéine amyloïdogène à l'aide d'anticorps spécifiques (figure 02.17)
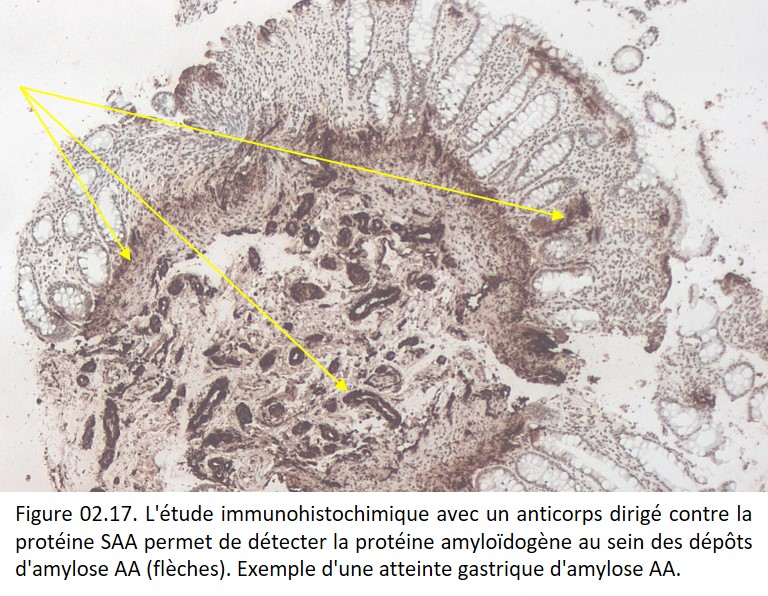{kind=link}
• La spectrométrie de masse après microdissection laser des dépôts : c'est maintenant la méthode de référence car elle permet de diagnostiquer tous les types d'amylose avec une grande fiabilité.
Traitements
Les modalités thérapeutiques dépendent de chaque type d'amylose mais ont un objectif commun : empêcher la synthèse de la protéine amyloïdogène soit en détruisant la cellule productrice comme dans l'amylose AL ou en stoppant spécifiquement la synthèse de la protéine comme dans l'amylose AA ou à ATTR.
Les formes localisées peuvent être traitées par exérèse locale mais elles récidivent souvent.
Calcifications
Causes
Les dépôts intratissulaires anormaux de calcium s'observent dans deux circonstances :
• calcifications dystrophiques, dans les tissus lésés, nécrosés, alors que la calcémie est normale ;
• calcifications dites métastatiques, dans les tissus sains à la faveur d'une élévation anormale de la calcémie.
Aspects macroscopiques
L'existence de calcifications se traduit macroscopiquement par une induration et une coloration blanc opaque, pierreuse (figure 02.18).
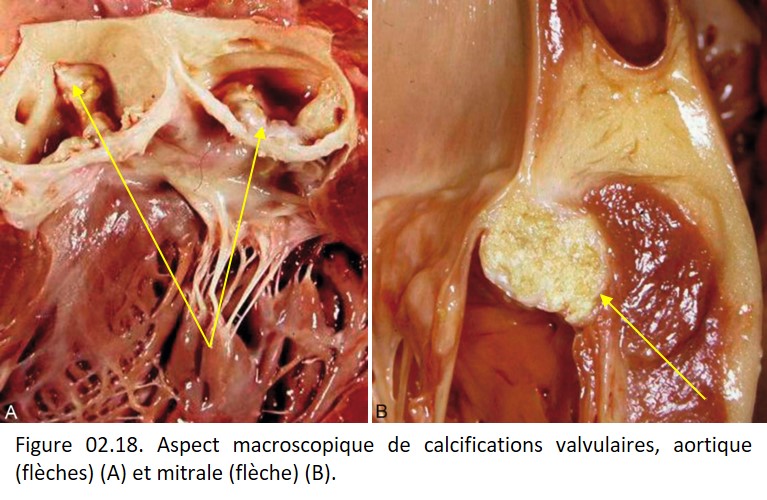{kind=link}
Divers aspects peuvent être réalisés, selon l'abondance et la distribution des précipités :
• « os de sèche » des pachypleurites ;
• « coque » des péricardites calcifiées ;
• « coquille d'œuf », dans l'athérosclérose calcifiée des gros vaisseaux ;
• « pierres », dans les fibromyomes utérins et les adénomes thyroïdiens involutifs calcifiés ;
• « craie » en cas de calcification partielle d'un tubercule caséeux ;
• « sable » dans le cas des sympexions prostatiques et des méningiomes psammomateux.
L'existence de ces calcifications est souvent bien mise en évidence par les radiographies.
Aspects histologiques
Sur une coloration par l'HES les dépôts calciques apparaissent denses, amorphes ou finement granulaires bleu-noir ou violacés (figures 02.19 et 02.20). Ils sont le plus souvent extracellulaires, plus rarement intracellulaires, et alors débutants, surtout observés en microscopie électronique, en particulier dans les mitochondries.
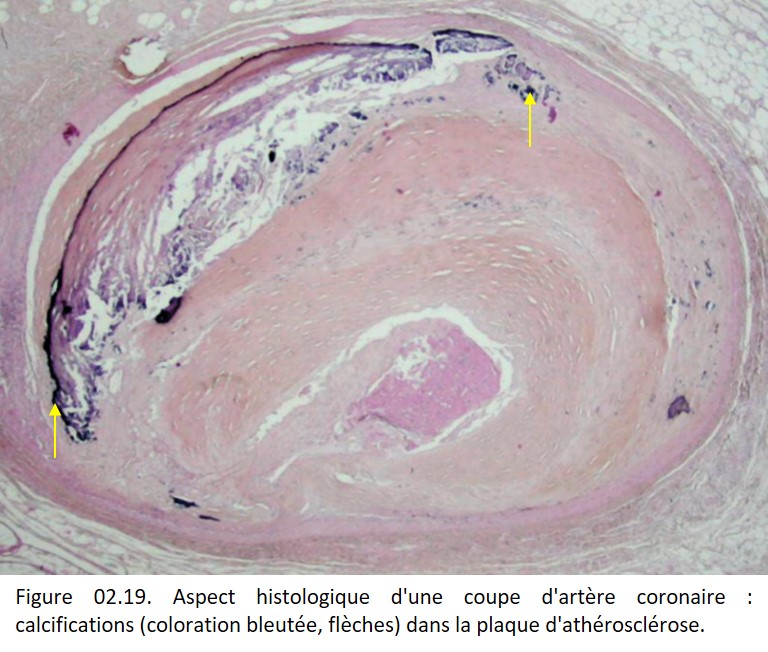{kind=link}
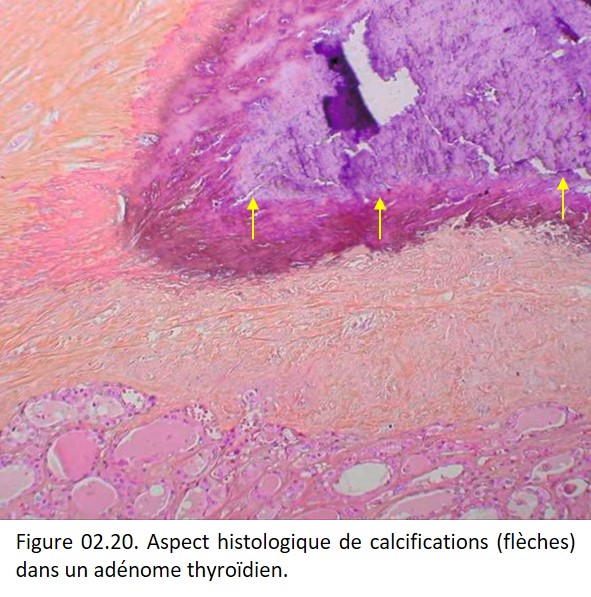{kind=link}
Ils peuvent s'accompagner d'une réaction macrophagique, type réaction à corps étrangers, voire d'une véritable ostéogénèse métaplasique.
Ils peuvent être mis en évidence par des colorations électives, comme le rouge d'alizarine et la réaction de Von Kossa.
Leur présence en abondance peut nécessiter une décalcification préalable du tissu (chélateurs de calcium, acide) avant l'inclusion en paraffine, ou bien l'utilisation de microtomes spéciaux pour objets durs.
Calcifications dystrophiques
Les calcifications peuvent survenir sur les tissus nécrosés, plus particulièrement dans la nécrose caséeuse de la tuberculose, la stéatonécrose, les infarctus anciens (myocarde), la « bouillie » athéromateuse (figure 02.19).
Elles peuvent aussi être favorisées par des altérations de la matrice conjonctive, comme dans la médiacalcose observée dans la média des artères au cours de la sénescence, les calcifications des tendons, de la dure-mère, des valves cardiaques (figure 02.18) ou du tissu sous-cutané.
Des calcosphérites, ou psammomes (calcification en strates concentriques) sont observées dans certaines tumeurs (méningiomes, carcinomes papillaires de la thyroïde ou de l'ovaire).
Des produits de sécrétion protéique accumulés dans des canaux peuvent servir de matrice aux dépôts de sels de calcium et donner naissance à des calculs (pancréas, voies biliaires).
La chondrocalcinose est à l'origine d'arthrites aiguës par précipitation de cristaux de pyrophosphate de calcium.
Calcifications métastatiques
Elles peuvent s'observer au cours d'hypervitaminose D, d'une ostéopathie destructrice (métastases osseuses, myélome), d'une hyperparathyroïdie primaire ou secondaire. Elles siègent dans le rein, au niveau de l'interstitium et des cellules tubulaires, dans les poumons, dans les cloisons interalvéolaires et dans les vaisseaux, le foie et le myocarde.
Dégénérescence et mort cellulaire
Lorsque les capacités d'adaptation d'une cellule sont dépassées, elle va s'engager dans un processus de dégénérescence (réversible) puis de mort cellulaire (irréversible). On distingue plusieurs types de mort cellulaire parmi lesquels la nécrose et l'apoptose sont les plus importants.
Les différentes cellules de l'organisme ont chacune des capacités d'adaptation propres aux différents types d'agression. Une agression systémique aura donc un retentissement variable en fonction des organes. Le retentissement sera également fonction de la durée et de la sévérité du stimulus délétère.
Si en pratique les agressions peuvent conduire à la dégénérescence puis à la mort cellulaire de plusieurs façons, celle-ci survient systématiquement en cas de lésions d'un des trois compartiments cellulaires suivants :
• mitochondries : elles sont indispensables pour maintenir la balance énergétique via la respiration cellulaire ;
• membranes plasmatiques : leur intégrité est nécessaire à la fois pour le maintien de l'homéostasie tissulaire et pour protéger la cellule des enzymes contenues dans les lysosomes ;
• matériel génétique : les cellules sont dotées d'un programme de surveillance qui déclenche une mort cellulaire dès qu'une lésion de l'ADN est mise en évidence.
Dégénérescence cellulaire
Définition
Il s'agit de l'ensemble des lésions élémentaires cellulaires, réversibles, pouvant précéder l'apparition de modifications cellulaires irréversibles correspondant à la nécrose.
Causes
Les causes sont les mêmes que pour la nécrose (développée dans la partie suivante).
Histologie
Les lésions dégénératives sont de deux types :
• dégénérescence hydropique (ou vacuolaire) : elle est caractérisée par un œdème intracellulaire qui se traduit par un gonflement de la cellule et un détachement apical de la membrane plasmatique en microscopie optique et par des anomalies ultrastructurales des mitochondries en microscopie électronique. Exemple : cellules épithéliales tubulaires rénales au cours de l'ischémie (figure 02.21) ;
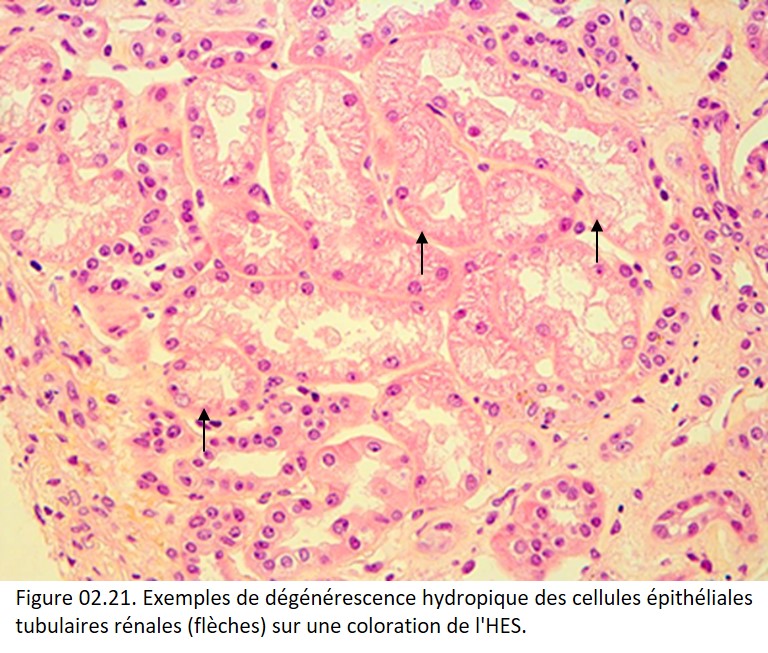{kind=link}
• dégénérescence graisseuse : elle est caractérisée par l'accumulation de triglycérides secondaire à la perturbation des voies métabolique des lipides. Exemple : lésions hépatocytaires dans la stéatose.
Nécrose cellulaire
Définition
La nécrose cellulaire désigne un type de mort cellulaire qui est caractérisé par la perte d'intégrité des membranes cytoplasmiques et par la libération des enzymes protéolytiques et des autres constituants de la cellule.
Attention : la nécrose doit être distinguée de l'autolyse qui survient à la mort de l'organisme.
Causes
Elles sont nombreuses et souvent entremêlées :
• anoxie, en particulier ischémie ;
• agents physiques, trauma mécanique, thermique, radiations ;
• agents chimiques et médicamenteux ;
• agents infectieux : virus, bactéries, champignons, parasites ;
• réactions immunologiques ;
• déséquilibres nutritionnels.
Histologie
Les modifications observables en microscopie optique traduisent la dénaturation protéique et la digestion des organites par les enzymes protéolytiques des lysosomes.
Attention : les modifications morphologiques associées à la nécrose ne sont visibles qu'au bout de plusieurs heures et il est donc possible de méconnaitre une nécrose très récente au microscope.
Nécrose cellulaire
Lésions élémentaires
• Le cytoplasme de la cellule nécrosée est habituellement éosinophile, par diminution de l'ARN cytoplasmique (responsable de la basophilie cytoplasmique) et par augmentation de la liaison de l'éosine aux protéines cytoplasmiques dénaturées (figure 02.22). Il peut être homogène ou vacuolaire (par digestion enzymatique des organites).
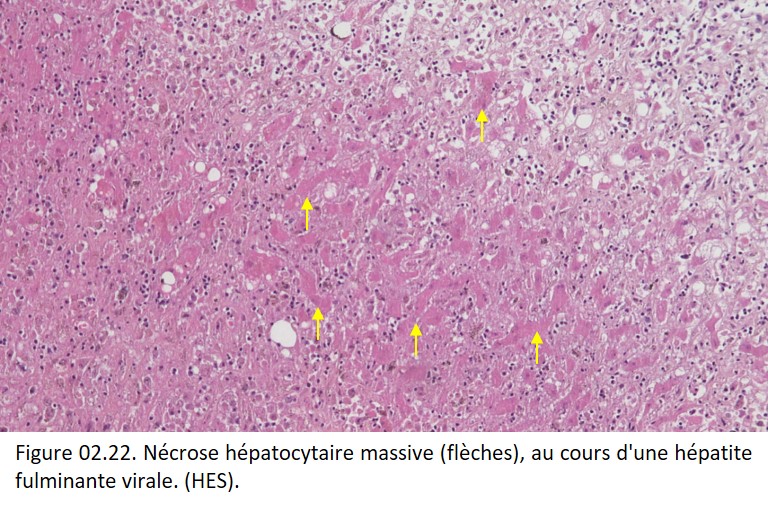{kind=link}
• Les modifications nucléaires sont constantes et prennent plusieurs formes :
pycnose : condensation avec rétraction du noyau et agglutination des amas chromatiniens contre la membrane nucléaire,
caryolyse : dissolution nucléaire avec perte des affinités tinctoriales,
caryorrhexis : fragmentation de la masse nucléaire.
Nécrose tissulaire
La nécrose cellulaire concerne habituellement des groupes de cellules dans un tissu, soumises aux mêmes agressions, par exemple lors d'un infarctus du myocarde après thrombose coronarienne, d'une nécrose œsophagienne après ingestion de caustiques, etc., et non pas des cellules isolées, comme pour l'apoptose.
Les différentes formes de nécrose
• Nécrose de coagulation, fréquente, lorsque la dénaturation protéique est l'événement essentiel, comme au cours de l'ischémie (figures 02.23 et 02. 24), des brûlures, de l'action de caustiques (figure 02.25). L'architecture tissulaire est préservée, fantomatique, les cytoplasmes sont éosinophiles et les noyaux pycnotiques ou en caryolyse.
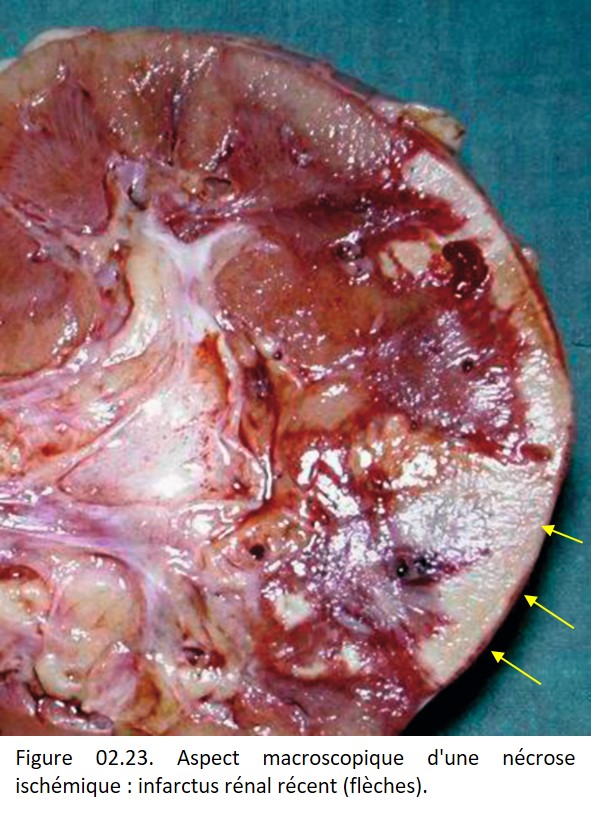{kind=link}
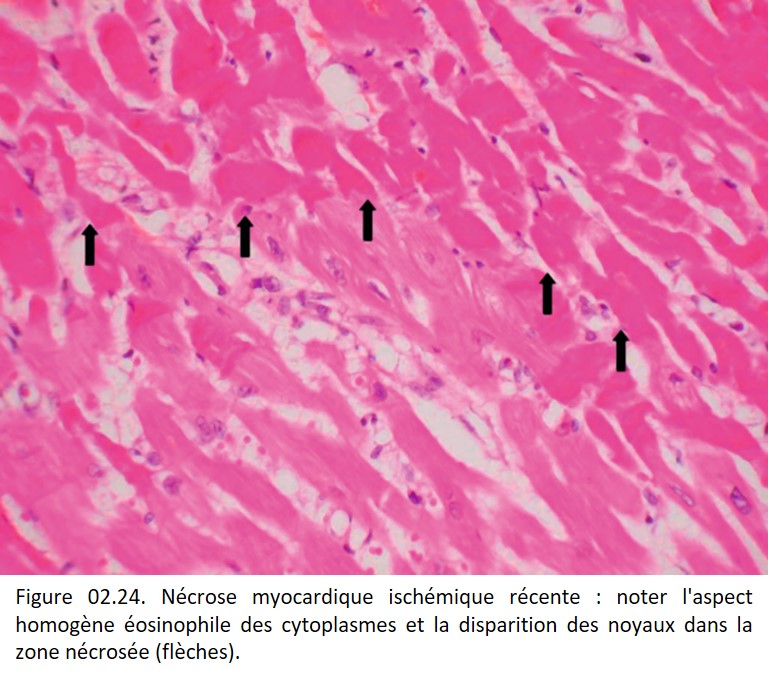{kind=link}
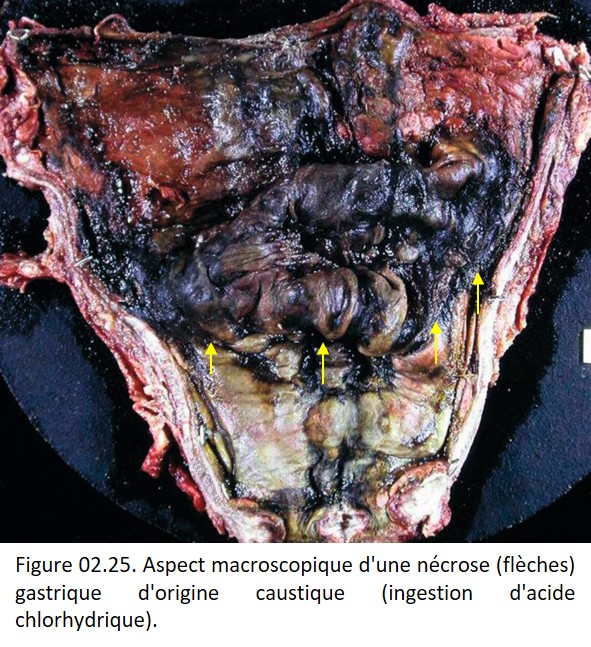{kind=link}
• Nécrose de liquéfaction, lorsque la digestion enzymatique domine, comme dans les infections à pyogènes. Elle comporte une perte totale de l'architecture tissulaire.
• Nécrose caséeuse, caractéristique de la tuberculose. Macroscopiquement, elle rappelle le lait caillé, d'où son nom de caséum (figure 02.26). Histologiquement, on observe un matériel nécrotique grumeleux, éosinophile, sans architecture cellulaire ou tissulaire (figure 02.27).
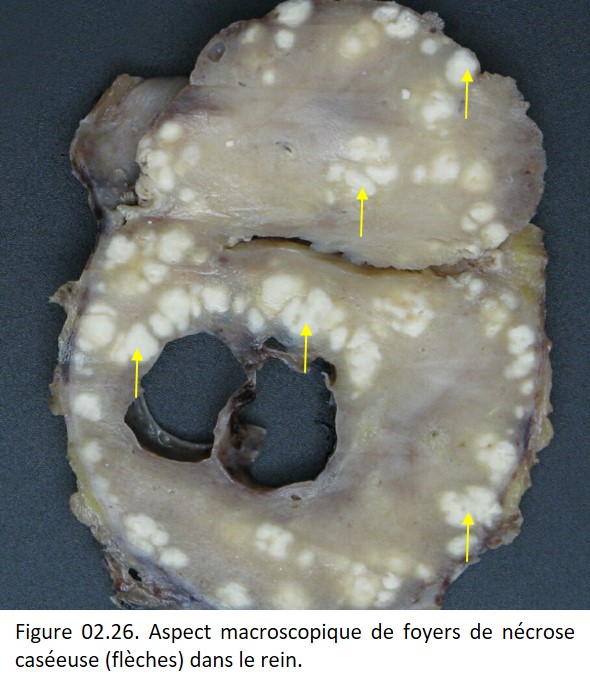{kind=link}
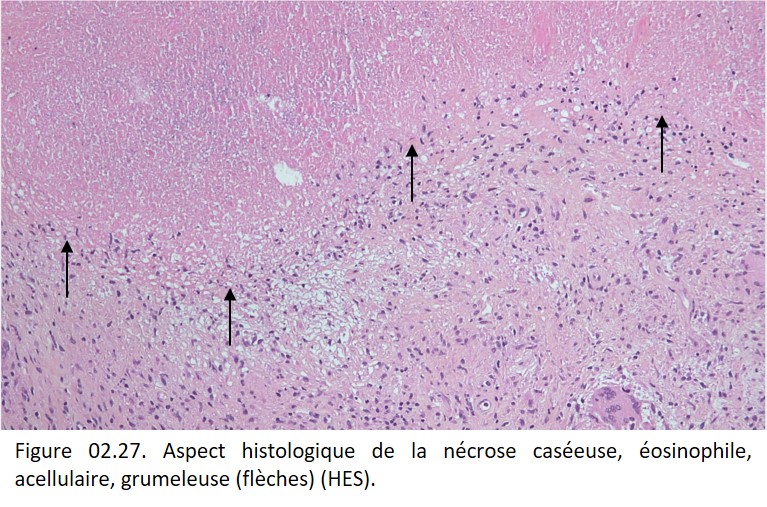{kind=link}
• Nécrose gangréneuse : elle est liée aux effets combinés de l'ischémie et de germes anaérobies.
• Cytostéatonécrose : c'est la nécrose du tissu adipeux (figure 02.28) qui est habituellement observée au cours de la pancréatite aiguë, par libération des enzymes pancréatiques lors de la nécrose du tissu exocrine (lipase). Macroscopiquement, la cytostéatonécrose a un aspect caractéristique crayeux, blanchâtre.
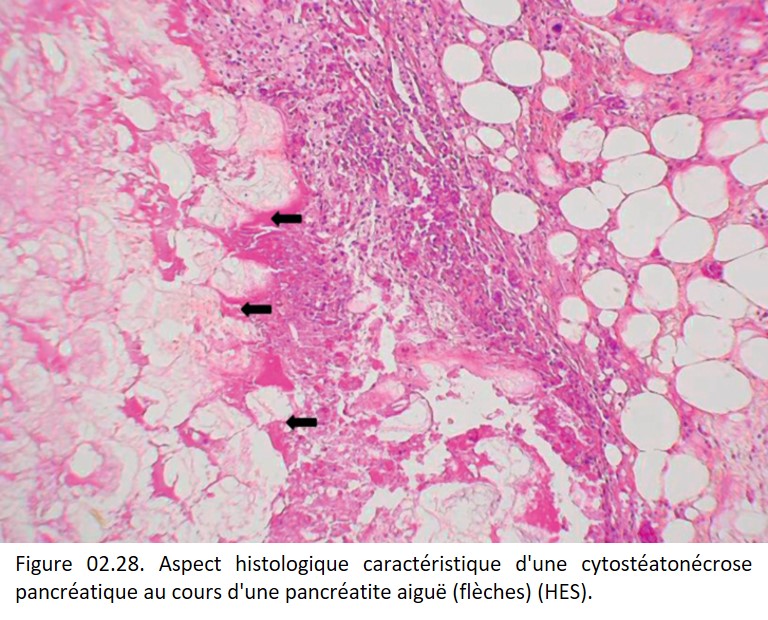{kind=link}
Évolution
Les cellules nécrotiques et leur contenu vont disparaitre secondairement à la digestion enzymatique et à la phagocytose des débris par les leucocytes (la nécrose entraine une inflammation). Si les cellules nécrotiques et les débris cellulaires ne sont pas rapidement détruits et réabsorbés, ils fournissent un nidus pour le dépôt de sels de calcium. La nécrose peut laisser place à une cicatrice. Si la nécrose est étendue, elle induit à sa périphérie une inflammation chronique qui peut évoluer progressivement en une coque fibreuse.
Apoptose
Définition
Nommée ainsi d'après le terme grec « tombé » elle fut individualisée en 1972 pour qualifier la mort cellulaire destinée à éliminer les cellules indésirables.
Il s'agit d'une mort cellulaire programmée qui est finement régulée. C'est un processus actif qui peut être déclenché par les cellules dans certaines conditions physiologiques ou en réponse à certaines lésions, en particulier aux dommages à l'ADN.
Elle concerne des cellules isolées, et non pas des groupes de cellules comme dans la nécrose.
Causes
On distingue les causes physiologiques d'apoptose des processus pathologiques.
Processus physiologiques
L'apoptose est un mécanisme physiologique de « suicide » cellulaire essentiel au développement, à la maturation, et au renouvellement normal des tissus. Elle intervient notamment :
• au cours de l'organogénèse (neurones) et de la croissance (involution thymique) ;
• au cours du développement de l'immunité (destruction des lymphocytes T autoréactifs) ;
• comme mécanisme d'homéostasie dans des tissus où le renouvellement cellulaire est permanent comme les cellules de l'épithélium gastro-intestinal, et les centres germinatifs des ganglions ;
• au cours de l'involution hormono-dépendante chez l'adulte : destruction des cellules endométriales au cours du cycle, régression des lobules mammaires après sevrage ;
• au cours du vieillissement.
Attention : l'apoptose étant un mécanisme important au cours du développement, un défaut d'apoptose peut être à l'origine d'une lésion (exemple : syndactylie membraneuse bilatérale et polydactylie (figure 02.29).
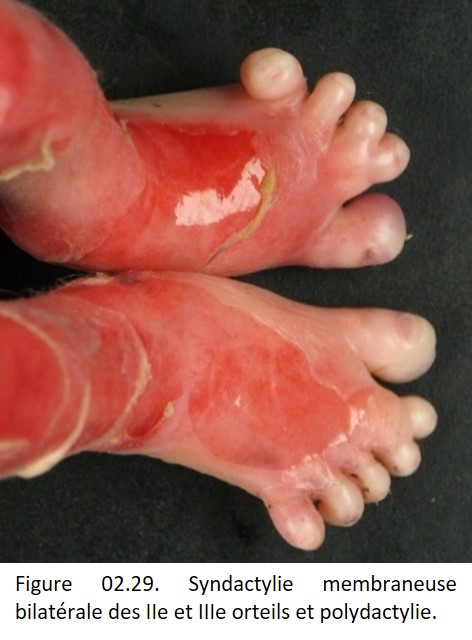{kind=link}
Processus pathologiques
Elle intéresse alors des cellules lésées ou des cellules reconnues comme étrangères ou tumorales par les lymphocytes T cytotoxiques ou NK, comme au cours du rejet de greffe, des hépatites virales.
Elle peut être induite par des stimuli qui à petites doses entraînent une apoptose, alors qu'à doses élevées ils induisent une nécrose : chaleur, irradiations, chimiothérapies anticancéreuses.
Elle est observée dans certains organes lors d'une obstruction canalaire, par exemple dans le pancréas, la parotide, le rein.
Physiopathologie
L'apoptose résulte classiquement de l'activation en cascade d'enzymes appelées caspases. Ces protéases sont normalement présentes dans une forme inactive dans la cellule et leur activation est donc un marqueur d'apoptose.
Deux voies distinctes convergent vers l'activation de ces caspases :
• la voie mitochondriale : le point de départ est l'augmentation de la perméabilité de la membrane externe mitochondriale. Elle est déclenchée par une perte de signaux de survie, des dommages à l'ADN ou encore l'accumulation de protéines mal repliées ;
• la voie des récepteurs de la mort : elle est déclenchée par l'engagement, à la surface des cellules, de récepteurs de mort par des ligands solubles ou membranaires. C'est par exemple le mécanisme de destruction cellulaire utilisé par les lymphocytes T cytotoxiques.
• Il existe toutefois des mécanismes d'apoptose indépendants des caspases.
Histologie
La cellule apoptotique apparaît en microscopie optique comme une cellule isolée des autres, rétractée, avec un cytoplasme éosinophile, comportant des fragments de chromatine nucléaire dense. À un stade débutant, la chromatine est condensée en périphérie du noyau. L'apoptose est quelquefois difficile à identifier car elle concerne des cellules isolées, ou très peu nombreuses, et n'induit pas de réaction inflammatoire (figure 02. 30).
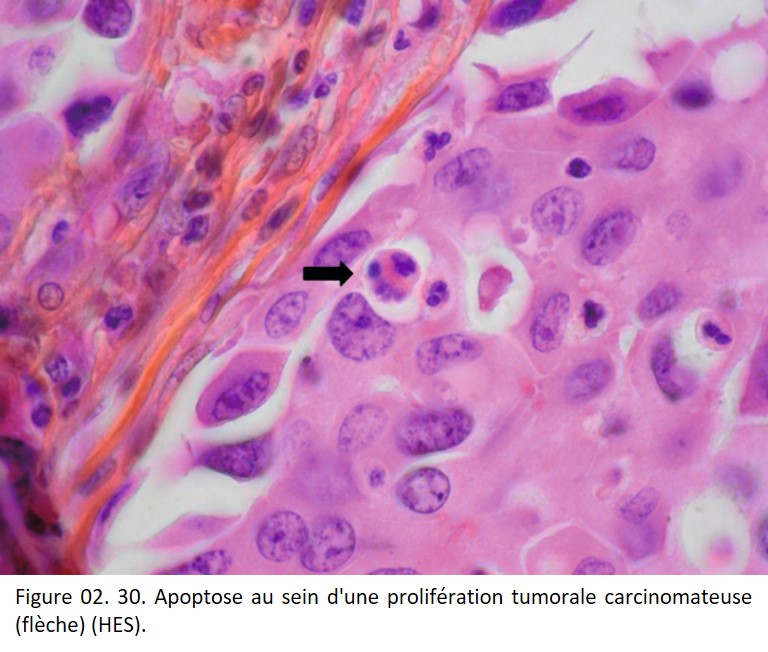{kind=link}
Les lésions sont mieux visibles, surtout aux stades précoces en microscopie électronique. La chromatine est condensée en périphérie du noyau, le nucléole est le siège d'une désintégration fibrillaire, des granulations osmiophiles apparaissent dans le nucléoplasme. Les organites intracytoplasmiques sont conservés, et les membranes restent très longtemps intactes, au contraire de la nécrose. Les structures de la surface cellulaire disparaissent, par exemple les micro-villosités de sorte que la cellule présente des contours lisses et s'isole des cellules voisines. Le volume cellulaire diminue. Finalement le noyau et la cellule elle-même se clivent en plusieurs fragments, entourés de membrane plasmique : ce sont les corps apoptotiques.
Plusieurs méthodes de détection in situ peuvent être utilisées : immunohistochimie, pour mettre en évidence la caspase 3 activée, et hybridation in situ pour la détection des terminaisons 3'OH au niveau des brisures internucléosomales de l'ADN (méthodes ISEL, In situ end labelling et TUNEL, TdT-mediated dUTP-biotin nick end labelling).
Évolution
Les cellules apoptotiques ainsi que les corps apoptotiques sont phagocytés par des macrophages ou par des cellules vivantes voisines. La cellule en apoptose est alors progressivement dégradée. Lorsque les cellules apoptotiques siègent dans un épithélium bordant une lumière elles peuvent aussi être éliminées dans la lumière (épithélium intestinal, etc.).
Différence avec la nécrose
L'apoptose et la nécrose s'opposent sur de nombreux points qui sont repris dans le tableau 02.01.
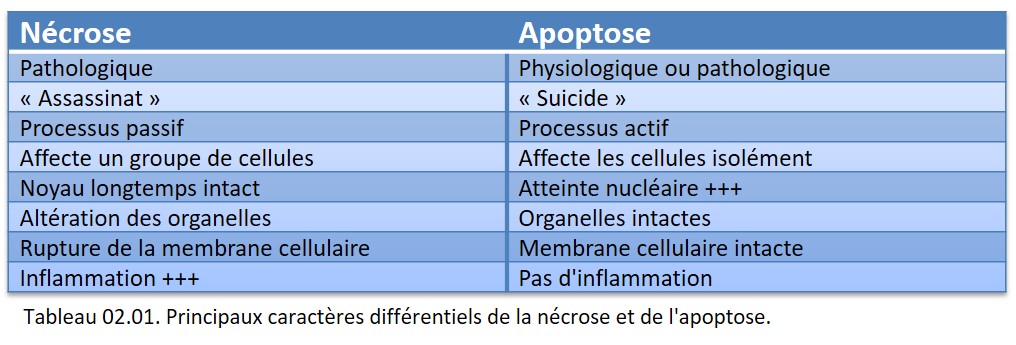{kind=link}
• L'homéostasie correspond au maintien de l'équilibre cellulaire et tissulaire. L'adaptation cellulaire permet à la cellule de trouver un nouvel équilibre lorsque son environnement est modifié par des exigences physiologiques plus importantes ou des circonstances pathologiques. Une lésion élémentaire est constituée par toute altération morphologique d'un élément vivant décelable par un quelconque moyen d'observation.
• Les principaux mécanismes d'adaptation cellulaire sont l'atrophie, l'hypotrophie, l'hypertrophie, l'aplasie, l'hypoplasie, l'hyperplasie et la métaplasie.
• Une terminologie spécifique à la pathologie du développement est utilisée pour décrire les lésions fœtales.
• Certaines lésions correspondent à l'apparition de substances intracellulaires, par exemple :
la stéatose qui correspond à l'accumulation anormale de triglycérides dans les cellules hépatocytaires,
la cholestase qui est définie histologiquement par une accumulation de bile dans le foie,
l'hémosidérose qui correspond à une surcharge tissulaire en hémosidérine, cette surcharge pouvant être localisée ou généralisée.
• Des substances exogènes peuvent s'accumuler dans les tissus, les cellules n'ayant pas le matériel enzymatique nécessaire à leur élimination. Elles peuvent être pourvoyeuses de maladie professionnelle comme dans la silicose.
• Certaines lésions touchent le compartiment intercellulaire. C'est par exemple le cas dans l'amylose qui correspond à un ensemble de maladies pouvant survenir dans des contextes très divers. Sa définition est histologique, directement secondaire aux propriétés tinctorielles des dépôts protéiques et à leur structure ultrastructurale particulière en feuillets β-plissés.
• Les calcifications correspondent à des dépôts intratissulaires anormaux de calcium, souvent visibles sur les radiographies. Les calcifications les plus fréquentes sont d'origine dystrophique, se formant dans des tissus préalablement lésés.
• Dans certains cas, l'agression est trop sévère et va dépasser les capacités adaptatives des cellules entrainant une dégénérescence puis une mort cellulaire.